

Ailevi Akdeniz Ateşi, tekrarlayan ateş, peritonit, sinovit, plörit, ve nadiren perikardit ve menenjit atakları ile karakterize, otozomal resesif geçişli genetik bir hastalıktır.
FMF Fenotip tip I, tekrarlayan ateş ve seröz zarların inflamasyonu (peritonitis, sinovitis veya plöritis) ile karakterize iken, fenotip II tanımı ise, tekrarlayan inflamasyon ve ateş öyküsü olmayan bir hastada, ilk bulgu olarak amiloidoz ortaya çıkması halinde kullanılmaktadır (1,2).
İnsidans
Hastalığın ülkemizde görülme sıklığı 1/1000 iken, taşıyıcı sıklığı 1/5 gibi oldukça yüksek bir orandadır. Bu durum, özellikle akraba evliliklerinin çok sık olduğu ülkemiz için ayrı bir öneme sahiptir. Akdeniz çevresindeki ırklar ve etnik gruplarda (Sefardik Museviler, Ermeniler, Türkler ve Araplar) nispeten sık görülmektedir (3).
Tanı Kriterleri
Ailevi Akdeniz Ateşi tanısı konmasında klinik bulgular ve aile öyküsü ile biyokimyasal ve genetik laboratuvar verileri kullanılmaktadır.
FMF’in klinik tablosu, abdominal ağrı (peritonit) ve/veya plöritik ağrı ve/veya artrit (ayak bileği ve diz) ile birlikte, 12-96 saat sürebilen tekrarlayan ateş atakları ile karakterizedir. Hastalık belirtilerinin belirli periyotlarla ortaya çıkması tanı için en önemli kriterdir.
Hasta eğer atak sırasında görülmüşse, atağa eşlik eden inflamatuvar bulgularının varlığı (lökositoz, sedimantasyon artışı, fibrinojen ve CRP’nin yükselmesi) ve bu testlerin atak sonlanınca normal değerlere inmesi tanıya yardımcı olur.
Bu testlerin pozitif bulunmasının FMF’e özgü olmadığı, sadece vücutta inflamasyonun varlığına işaret ettiği unutulmamalıdır.
Klinik pratikte, hastalığın tanısı için en sık Tel-Hashomer kriterleri kullanılmaktadır(4).
| Tablo. Tel-Hashomer Kriterleri |
|---|
| Majör kriterler |
| • Poliserözit ile seyreden tekrarlayan ateş atakları • Başka bir nedene bağlanamayan AA tipi amiloidoz • Sürekli kolşisin tedavisine iyi yanıt |
| Minör kriterler |
| • Yineleyen ateşli ataklar • Erizipel benzeri döküntü • Birinci derece akrabada FMF varlığı |
| Kesin tanı : 2 majör veya 1 majör + 2 minör kriter Olası tanı : 1 majör + 1 minör kriter |
| FMF |
|---|
 |
Aile öyküsünün pozitif olması tanıyı destekleyen önemli bir bulgudur. Bununla beraber, FMF’in resesif geçişli bir genetik hastalık olması nedeniyle, ailede başka bir indeks vaka bulunmayabileceği de unutulmamalıdır.
Hastalığın Kalıtım Modeli ve Moleküler Genetiği
FMF otozomal resesif geçişli bir hastalıktır, ancak literatürlerde az sayıda otozomal dominant geçişli vakalar da bildirilmiştir. FMF’den sorumlu olan MEFV geni, kromozom 16p13.3’de lokalizedir, 10 ekzondan oluşur ve Pyrin proteinini kodlar (Tablo 1).
Pyrin proteini, başlıca nötrofil ve monositlerde sentezlenir ve kaspaz-1 ile interlökin-1β aracılığıyla apoptozda görev alacak proteinlerin ekspresyonunu ve anti-inflamatuvar aktivitelerini düzenler.
MEFV geninde oluşan mutasyonlar, pyrin ekspresyonunu azaltır. Buna bağlı olarak da interlökin-1β aktivasyonunu sağlayan kaspaz-1 üzerindeki etkinin azalması sonucunda, proteinin inflamasyondaki kontrol görevinin aksamasıyla, uyarılmış olan inflamasyon durdurulamaz ve ateşle birlikte belirli bölgelere sınırlı inflamasyon atakları şeklinde klinik tablo ortaya çıkar.
FMF’li Hastalar Şekil: 2 |
Hastalığa neden olan mutasyonlar, en fazla genin 2. ve 10. ekzonlarında bulunur. En sık gözlenen mutasyonların gen içerisindeki dağılımları tablo 2’de görülmektedir.
Tablo 2: MEFV genindeki mutasyonların dağılımları Tablo 2: MEFV genindeki mutasyonların dağılımları |
Genetik Tanı Endikasyonları
FMF hastalığında genetik tanı, klinik tanının doğrulanması, sınırda vakalarda tanının kesinleştirilmesi ve taşıyıcıların saptanması amacıyla yapılmaktadır. Prenatal amaçlı olarak da yapılabilir.
Türk FMF Çalışma Grubu’nun 35 merkezde 2838 vakalık çok merkezli çalışmasında, Türk popülasyonunda FMF hastalığının başlangıç yaşının ortalama 9.6, tanı koyma yaşının ise 16.4 olduğu tespit edilmiştir.
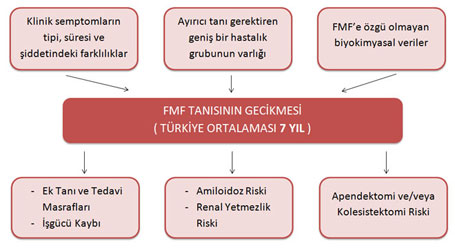
Bu çalışmanın en çarpıcı sonuçlarından birisi de, ekonomimize ciddi yükler (tanı ve tedavi masrafları, iş gücü kayıpları, amiloidoz ve böbrek yetmezliği komplikasyonları) getiren bu genetik hastalığın tanısının yaklaşık 7 yıl gecikme ile konulabiliyor olmasıdır.
Yine, aynı literatürde, FMF tanısı ile sonuçlanan bu uzun süreçte vakaların yaklaşık % 19.2’sinin apendektomi ve % 1.9’unun da kolesistektomi operasyonu geçirmiş olduğu bildirilmektedir(5,6).
FMF hastalığına neden olan MEFV genindeki mutasyonların tanı değerleri ve kullanılan moleküler teknikler tablo 3’de görülmektedir.
Tablo 3. Test metodolojisi ve tanı değeri
| Yöntem | Test adı | Tanı değeri |
| PCR / Reverse Hibridizasyon | 12 Mutasyon M694V, M680I(G/C), M680I(G/A), E148Q, V726A,M694I, P369S, F479L, K695R, A744S, R761H, 1692 del | %85 |
| PCR / Dizi Analizi | 40 Mutasyon | %85-90 |
| PCR, Dizi Analizi (Tüm ekzonlar) | Tüm gen analizi | ≥%90 |
FMF araştırmasında klinik değerlendirmeyi takiben moleküler genetik incelemenin yapılması çok önemlidir.
Klinisyen, mutasyonlar ile klinik semptom karşılaştırmasını yaparak, bazı mutasyon tiplerinin heterozigot formda olsa dahi, klinik semptom verebileceğini göz önünde bulundurmalıdır.
 FMF tanısının gecikmesi |
Özellikle homozigot olmak üzere M694V mutasyonunu taşıyan Kuzey Afrikalı Yahudilerde %90 olguda mutasyonla amiloidoz gelişmesi arasında sıkı bir ilişki gösterilmiştir. Bazı çalışmalarda M694V mutasyonu ile ağır bir fenotip bildirilmesine rağmen, bu bulguyu desteklemeyen çalışmalar da mevcuttur(7-10). Bununla birlikte aile içi ve aileler arası klinik farklılıklara dayanarak fenotipin, MEFV lokusu dışındaki genler ve/veya çevresel etmenlerden etkilendiği düşünülmektedir.
Tedavi
FMF önlenebilir ve kontrol edilebilir bir hastalıktır. Hastalığın oluşturduğu klinik tabloyu düzeltme amacıyla çiğdem bitkisinden elde edilen kolşisin kullanılmaktadır. Kolşisin, FMF tedavisinde 2 önemli amaçla kullanılır.
1. Atakların engellenmesi veya hafifletilmesi
Düzenli olarak kolşisin kullanan hastalarda ataklar ya hiç tekrarlamaz ya da öncekilere oranla çok daha seyrek hale gelir ve hafif geçerler.
Sadece atak döneminde kullanılmasının bir yararı yoktur ve bu şekilde başlamış olan atağa önleyici bir etki sağlamaz. Etkinliği ilacın düzenli kullanımına bağlıdır.
2. Amiloidoz gelişiminin engellenmesi
Kolşisin, hekim kontrolünde düzenli ve yeterli dozda kullanıldığında amiloidoz gelişimini engellemektedir. M694V mutasyonunu taşıyan kişilerin amiloidoz gelişimi için daha yüksek riske sahip oldukları ve bu nedenle mutlaka kolşisin kullanımı gerektiği bildirilmektedir. Tedavi takibinde gerekli hassasiyetin gösterilmemesi durumlarında klinik tablo tekrar bozulabilmektedir.
Genetik Danışma
FMF, otozomal resesif kalıtım göstermektedir. Hasta bireyin anne ve babası zorunlu taşıyıcıdır. FMF taşıyıcılığı ve akraba evliliği oranı yüksek olan toplumlarda çocukların taşıyıcı veya hasta olarak dünyaya gelme olasılığı yüksektir. Eğer anne ve baba mutasyonu heterozigot olarak taşıyorsa bir sonraki nesilde hasta bireyin dünyaya gelme ihtimali %25, taşıyıcılık oranı %50 iken sağlıklı birey olma olasılığı %25’dir.
Ailesel Akdeniz Ateşi, hekimlerin periyodik karın ağrısı, göğüs ağrısı, ateş ve artrit şikâyetleri ile tanıda zorlandığı bir hastalıktır. FMF ile benzer semptomlar gösteren hastalıkların ayırıcı tanısının güçlüğü göz önüne alındığında, moleküler tanı metotları önem kazanmıştır. Tüm genetik hastalıklarda olduğu gibi, FMF için de genetik tanı uygulaması yapılmalı ve danışma hizmeti verilmelidir. Her hastanın taşıdığı mutasyon tespit edilerek “genetik kimlik kartı” alması sağlanmalıdır.
Hasta oranının 1:1000 ve taşıyıcı oranın 1:5 olduğu ülkemizde, FMF kaynaklı ciddi bir sağlık sorunu bulunmaktadır. Hastalarımızın FMF’in genetik boyutu hakkında ciddi şekilde bilgilendirilmesi ile sonraki jenerasyonlarda maddi-manevi kayıpların azaltılması hedeflenmelidir.
Sık Sorulan Sorular
1. Ataklar FMF’de nasıl seyreder?
FMF atakları tekrarlayan ateş ve ağrılı durumlardır ve önceden kestirilmesi zordur. Ataklar genelde 2-3 gün sürer ve en şiddetli atak ilk 12-24 saatte görülür.
2. FMF tanı kriterleri nelerdir?
• 38-40 derece arası yüksek ateş
• Tekrarlayan karın ağrısı
• Tekrarlayan göğüs ağrısı
• Ağrılı ve şiş eklemler
• Kabızlığı takip eden ishal
• Bacaklarda özellikle diz altlarında kırmızı döküntüler
• Nadiren kas ağrıları, kadınlarda üreme organları iltihabı, erkelerde şiş ve hassas testisler ve vaskülit (damar iltihabı)
3. Laboratuvar testleri FMF’in kesin tanısı için yeterli midir?
Hayır, sadece FMF ‘e özgü bir laboratuar testi yoktur. Tanı konulmasında diğer hastalıkların elenmesi, aile hikayesi, Tel-Hashomer kriterleri, ataklar sırasında yükselen inflamatuvar parametrelerin laboratuvar testleri ile gösterilmesi esas alınır. Klinik bulgularla tanı konulmasına rağmen kesin tanı için MEFV genindeki mutasyonların genetik test ile taranması önerilir. Hastalığa ülkemizde yaygın olarak kullanılan testler ile (strip test ve ekzon 2-10 dizi analizi) %85’lik tüm gen analizi(10 ekzon) ile %90’dan daha fazla oranda tanı konulabilmektedir. Bu genetik testler laboratuvarımızda yapılabilmektedir.
4. FMF’in ciddi komplikasyonları var mıdır?
Evet. Eğer hastalığın tanı ve tedavisi gecikirse ciddi komplikasyonlar oluşabilir.
Amiloidoz en sık rastlanan komplikasyon olup böbreklerde Amiloid A denilen bir proteinin birikmesine yol açar. Bu birikme idrarda aşırı protein kaybı ile karakterize edilen nefrotik sendroma ve daha sonra renal yetmezliğe sebep olur. En sık görülen M694V mutasyonunda amiloidoz riski çok yüksektir. Bu sebeple amiloidoz riskinin önceden belirlenebilmesi için genetik test yapılması ve hastaya mutasyon kaynaklı risk bilgisinin genetik danışma ile verilmesi önerilmektedir. FMF üreme organlarında inflamasyona yol açtığında infertilite görülebilir. FMF’in neden olduğu diğer bir hastalık olarak kronik artrit; eklemler, diz, ayak bileği, kalça ve dirsekte görülen ağrılar ile seyireden bir hastalıktır ve eklem tahribatı olmadan iyileşebilir. Kesin FMF tanısındaki gecikme ile beraber hastalığın seyri yaşam kalitesini düşürebilir.
Referanslar:
1. Shinar Y, Livneh A, Langevitz P, Zaks N, Aksentijevich I, Koziol DE, Kastner DL, Pras M, Pras E.Genotype-phenotype assessment of common genotypes among patients with familial Mediterranean fever. . J Rheumatol. 2000;27(7):1703-7.
2. Shohat M, Magal N, Shohat T, Chen X, Dagan T, Mimouni A, Danon Y, Lotan R, Ogur G, Sirin A, Schlezinger M, Halpern GJ, Schwabe A, Kastner D, Rotter JI, Fischel-Ghodsian N. Phenotype-genotype correlation in familial Mediterranean fever: evidencefor an association between Met694Val and amyloidosis. Eur. J. Hum. Genet. 1999;7(3):287-92. 32.
3. Sohar E, Gafni J, Pras M, et al. Familial Meditarrenean fever: a survey of 470 cases and review of the literature. Am J Med 1967; 43:227-253.
4. Livneh A, Langevitz P, Zemer D, et al. Criteria for the diagnosis of familial Mediterranean fever. Arthritis Rheum 1997;40(10):1879-85.
5. O. Sayarlioglu M, Cefle A, Inanc M, Kamali S, Dalkilic E, Gul A, Ocal L, Aral O, Konice M. Characteristics of patients with adult-onset familial Mediterranean fever in Turkey: analysis of 401 cases. Int J Clin Pract. 2005;59(2):202-5.
6. Turkish FMF Study Group. January 2005. Familial Mediterranean Fever in Turkey. Results of a Nationwide Multicenter Study. Medicine; 84:1-11
7. Pras E, Langewitz P, Livneh A, Zemer D, Migdal A, Padeh S, Lubetzky A, Aksentijevich I, Centola M, Zaks N, Deng Z, Sood R, Kastner DL, Pras M. Genotype/phenotype correlation in familial Mediterranean fever (a preliminary report). In: Sohar E, Gafni J, Pras M (eds) Familial Mediterranean Fever. Freund Publishing House, Tel Aviv, pp 260-4. 1997
8. Shinar Y, Livneh A, Langevitz P, Zaks N, Aksentijevich I, Koziol DE, Kastner DL, Pras M, Pras E. Genotype-phenotype assessment of common genotypes among patients with familial Mediterranean fever. J Rheumatol. 2000;27:1703–7.
9. Shohat M, Magal N, Shohat T, Chen X, Dagan T, Mimouni A, Danon Y, Lotan R, Ogur G, Sirin A, Schlezinger M, Halpern GJ, Schwabe A, Kastner D, Rotter JI, Fischel-Ghodsian N. Phenotypegenotype correlation in familial Mediterranean fever: evidence for an association between Met694Val and amyloidosis. Eur J Hum, Genet. 1999;7:287–92.
10. Ozcakar ZB, Yalcinkaya F, Yüksel S, et al. Possible effect of subclinicalinflammation on daily life in familial Mediterranean fever. Clin Rheumatol 2006;25:149-52.
Son Güncelleme 20 Şubat 2023